把不确定性前置:现代药物发现的真正一体化
现代药物发现的让人眼花缭乱:HTS、DEL、FBDD、virtual screening、ASMS、SPR、Cryo-EM、AIDD、DMPK、PK/PD、tox、CMC。每一种技术都有自己的拥护者,也都有足够多的成功案例可以证明自己重要。
现代药物发现的让人眼花缭乱:HTS、DEL、FBDD、virtual screening、ASMS、SPR、Cryo-EM、AIDD、DMPK、PK/PD、tox、CMC。每一种技术都有自己的拥护者,也都有足够多的成功案例可以证明自己重要。
但真正让项目变慢、变贵、变混乱的,往往不是技术不够,而是问题没有被放在正确的位置上。一个靶点还没有被证明可筛,团队已经在争论用哪种库;一个 primary hit 还没有排除假阳性,项目已经开始讲 SAR;一个分子只有体外活性,团队已经把它想象成候选物;一个项目连续失败,却因为“还有一个想法”迟迟没有停下来。
这场关于 integrated screening and chemistry 的圆桌,最有价值的地方不在于推荐某一个平台,而在于给出了一套更朴素、也更接近真实研发的判断方式:先判断不确定性在哪里,再决定用什么技术购买下一块证据。
一体化不是能力清单,而是时间轴
很多公司都会说自己有一体化平台。问题是,一体化如果只是一张能力清单,其实意义有限。真正有用的一体化,应该是一条从 target 到 commercial 的时间轴:早期有 protein production、structural biology、membrane protein、Cryo-EM、biophysical technology 和 hit discovery;中段有 bioassay、AIDD/CADD、medicinal chemistry、pharmacology 与 DMPK;再往后要接上 CMC、API、formulation、scale-up 和 manufacturing。
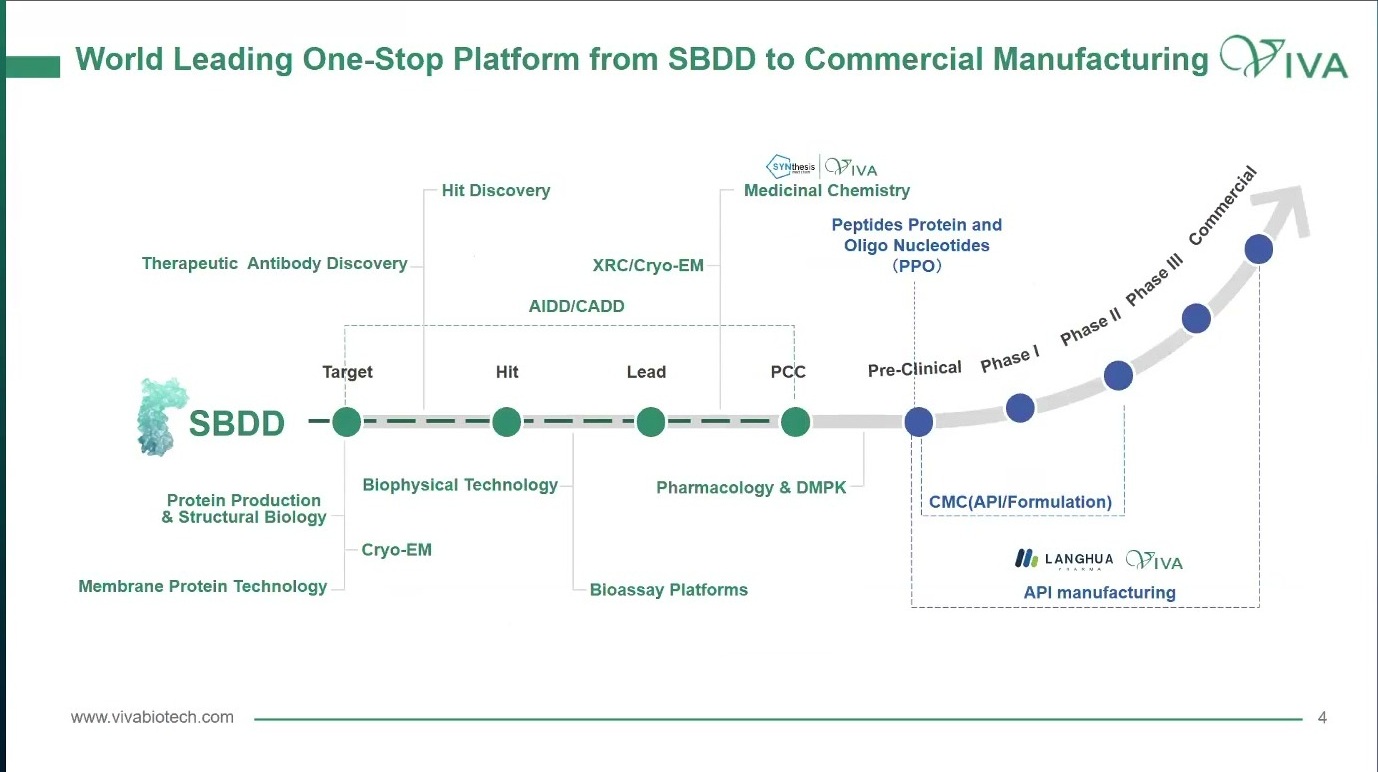
图1. 一体化平台如果只是一张能力清单,价值有限;关键是它是否能沿着 target、hit、lead、PCC 到 commercial 的时间轴传递问题。
这条时间轴提醒我们:药物发现不是把多个部门并排放在一起,而是让每个阶段的问题能够自然传到下一个阶段。
Target 阶段要回答的是“这个靶点是否值得投入”。Hit 阶段要回答的是“这个信号是不是假象”。Lead 阶段要回答的是“这个系列能不能被优化”。PCC 阶段要回答的是“这个分子能否在药效、暴露、安全性、可开发性之间同时站住”。如果这些问题没有被按顺序组织起来,再强的平台也会变成局部最优的堆叠。
AI 不是跳过实验的捷径,而是嵌入这个时间轴的加速器:在 modeling 阶段帮助结构预测、binding site prediction 和 virtual screening;在 generation 阶段做 de novo design、hit selection 和属性优化;在 validation 阶段协助检测方法、合成测试和模型优化;在 lead optimization 阶段参与 FEP、MD 和多目标优化。AI 真正的价值不在于“替代研发”,而在于让下一轮实验更值得做。
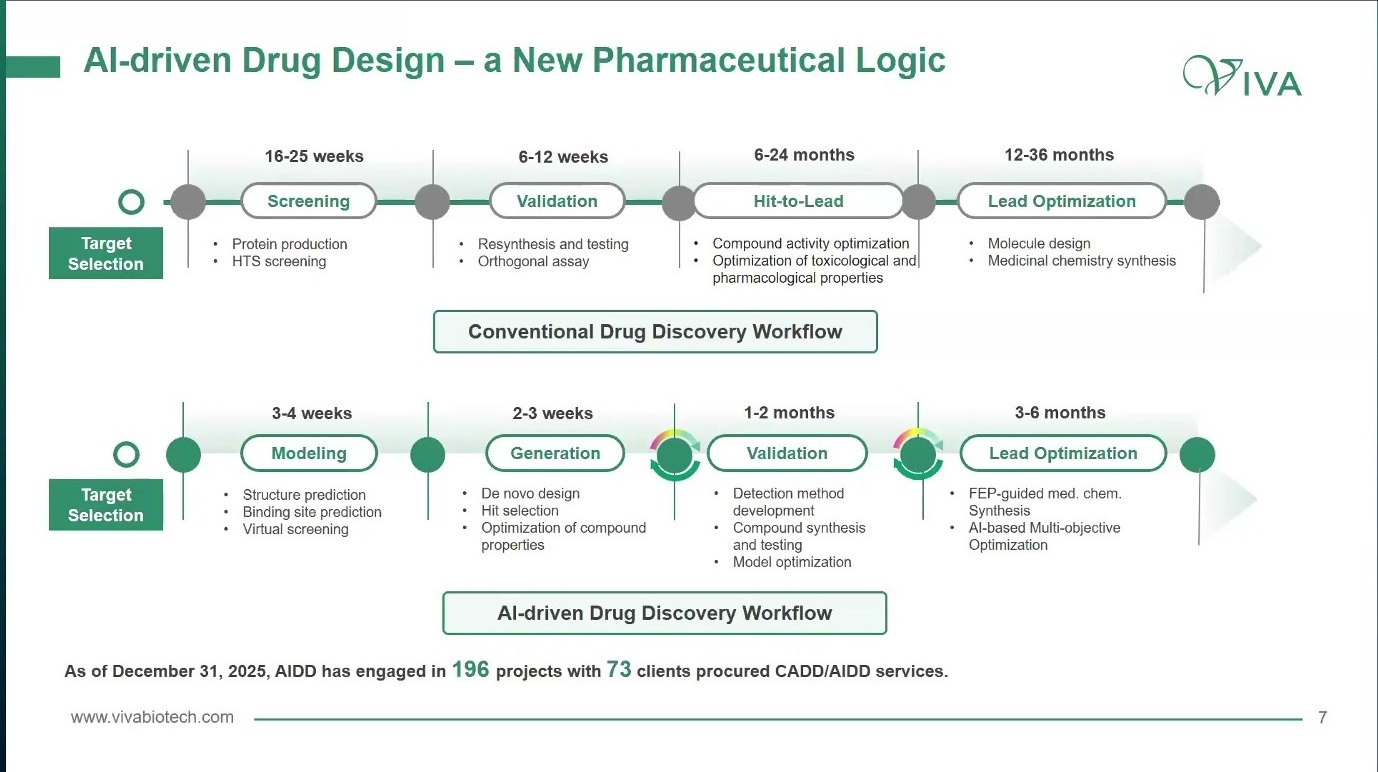
图2. AI-driven workflow 的位置不是替代实验,而是把 modeling、generation、validation 和 lead optimization 连接到下一轮可执行实验。
所以,一体化的第一层含义不是“我有多少平台”,而是“我能否让每个平台在正确的时间回答正确的问题”。
Hit ID 的第一问,不是“用 HTS 还是 DEL”
早期项目最容易犯的错误,是把技术平台当作答案。手里有 HTS,就想把问题变成 HTS;熟悉 DEL,就倾向于把问题变成 DEL;有结构信息,就自然想从 docking、FBDD 或 virtual screening 切入。
但更好的顺序应该反过来:先问靶点,再问 assay,再问材料和资源,最后才问平台。

图3. Hit ID 不是平台偏好题,而是结构信息、assay 可用性、蛋白量、化学空间、预算与基础设施共同决定的证据购买路径。
如果一个靶点已经有可靠的 3D structure,例如 X-ray、NMR 或 Cryo-EM,那么 FBDD 和 virtual screening 就有了更好的起点。结构信息能把化学空间收束到更有意义的位置,fragment 也更容易被解释和扩展。
如果没有结构,但已经有可靠的 functional 或 cellular assay,HTS 的价值会变高。它能直接验证生物活性,尤其适合资源和基础设施允许、且需要在大规模物理库中寻找起点的项目。
如果 functional assay 还不可靠,或者细胞体系太复杂,那么 affinity-first 的思路更稳。ASMS、SPR、DEL 等方法至少可以先回答一个更底层的问题:分子是否真的接触靶点。没有这个层面的证据,后面的功能 readout 很容易被系统干扰带偏。
如果蛋白极难表达、产量极低,而项目又需要探索巨大的化学空间,DEL 的吸引力就来自它对蛋白量的要求较低,以及用 DNA 编码把超大库压进一个可筛选格式的能力。相反,如果蛋白充足、结构明确、问题聚焦,ASMS、SPR、FBDD 或更小而精的物理库也许更直接。
预算和基础设施同样会改变答案。高资本投入可以支撑 HTS、自动化液体处理、物理库维护和高通量检测;资源更紧的团队,则可能更需要 virtual screening、DEL 或小规模快速评估来先购买关键信息。
这套决策树的核心不是“哪种平台更高级”,而是“当前最缺哪种证据”。强 genetic validation、明确 biological rationale、较好 tractability 的靶点,可以并行多平台,以提高获得多样起点的概率。高风险、低表征的靶点,则更适合先做小规模或快速评估:先确认小分子或肽路线是否有机会,再决定要不要扩大投入。
这就是复杂性管理的第一条规则:不要在靶点问题还没定义清楚时,就过早选择技术答案。
Modality 选择要前置,否则后面会很贵
很多项目把 modality 当成后置选择:先找 hit,找到了再看它能不能发展成小分子、肽、抗体或其他形式。但对难靶点和 first-in-class 靶点来说,这个顺序常常太晚。
更合理的方式,是在 target recommendation 之后就做 ligandability 和 modality assessment。遗传数据、gene mapping、AI data mining 可以帮助形成靶点优先级;随后要问的是:这个靶点更像 small molecule target,还是更适合 linear/cyclic peptide?如果这些路线不成立,是否应该转向 antibody、oligonucleotide、cell therapy 或其他 modality?
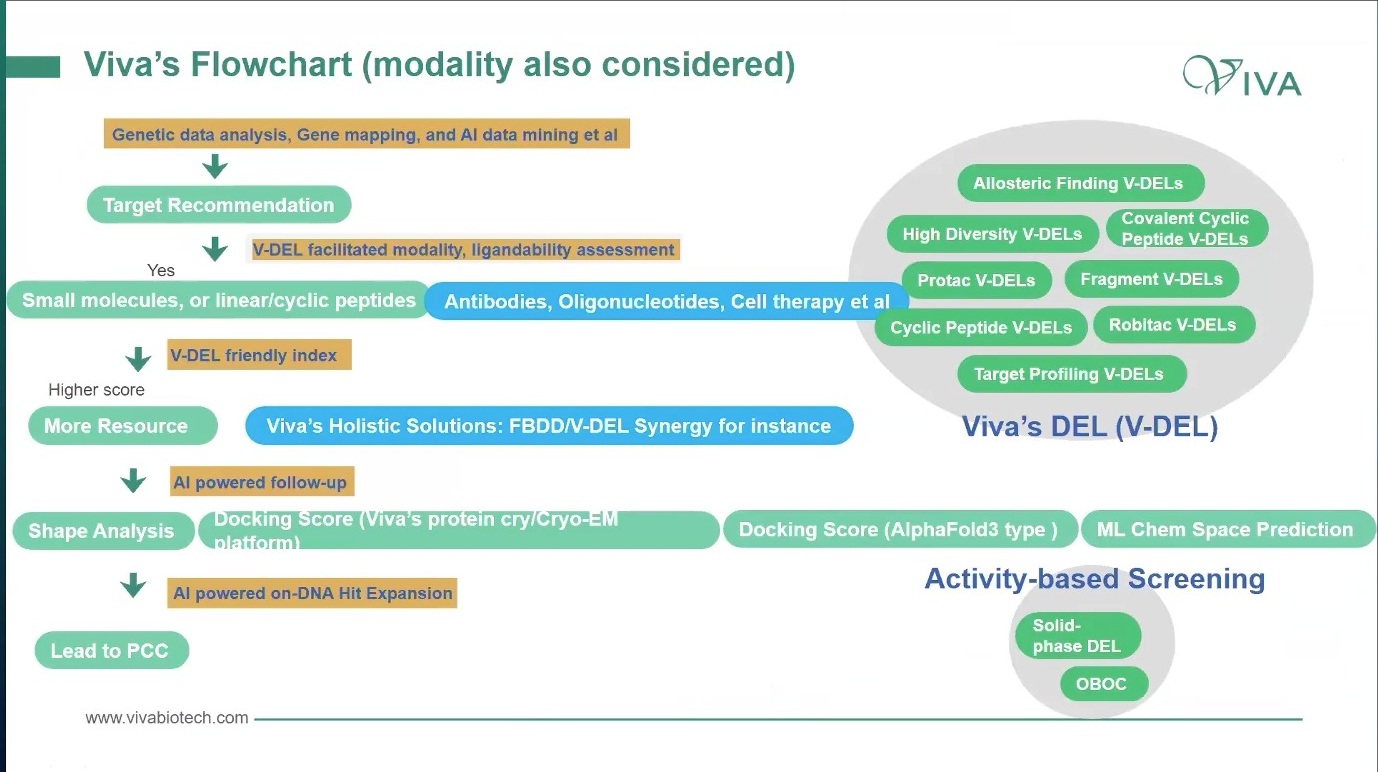
图4. Modality 需要在 target recommendation 后前置评估:先判断靶点和配体可行性,再决定小分子、肽、DEL/V-DEL 或其他治疗形式。
DEL 或 V-DEL 在这里的角色就不只是“找 hit”。它还可以作为 target triage 工具:用较少蛋白和较大化学空间,快速观察某些靶点是否产生可解释的富集、是否有 ligandability 信号、是否出现可扩展的 active series。再结合 post-selection chemistry、分子特征、蛋白结构或 Cryo-EM 信息,可以形成 target / modality / platform priority index。
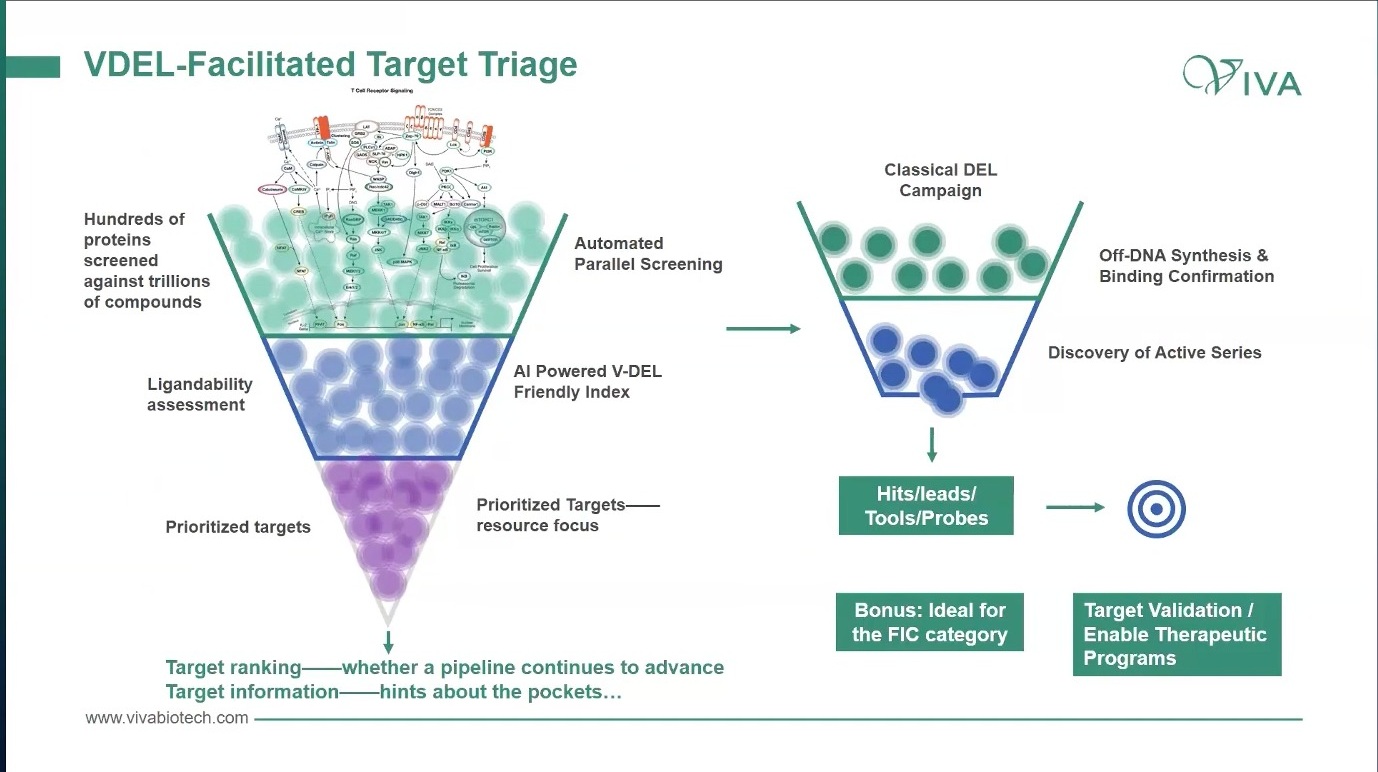
图5. V-DEL 可以被用作 target triage:用平行筛选和 ligandability 观察,把靶点排序和资源聚焦提前。
这里必须保持克制:这种指数或算法不能因为出现在流程图里,就被当成已经独立验证过的行业标准。它更像一种资源分配工具。它的意义在于把原本凭经验争论的事情,尽量变成数据化排序:哪个靶点更值得继续,哪种 modality 更值得推进,哪种平台更可能给出下一步答案。
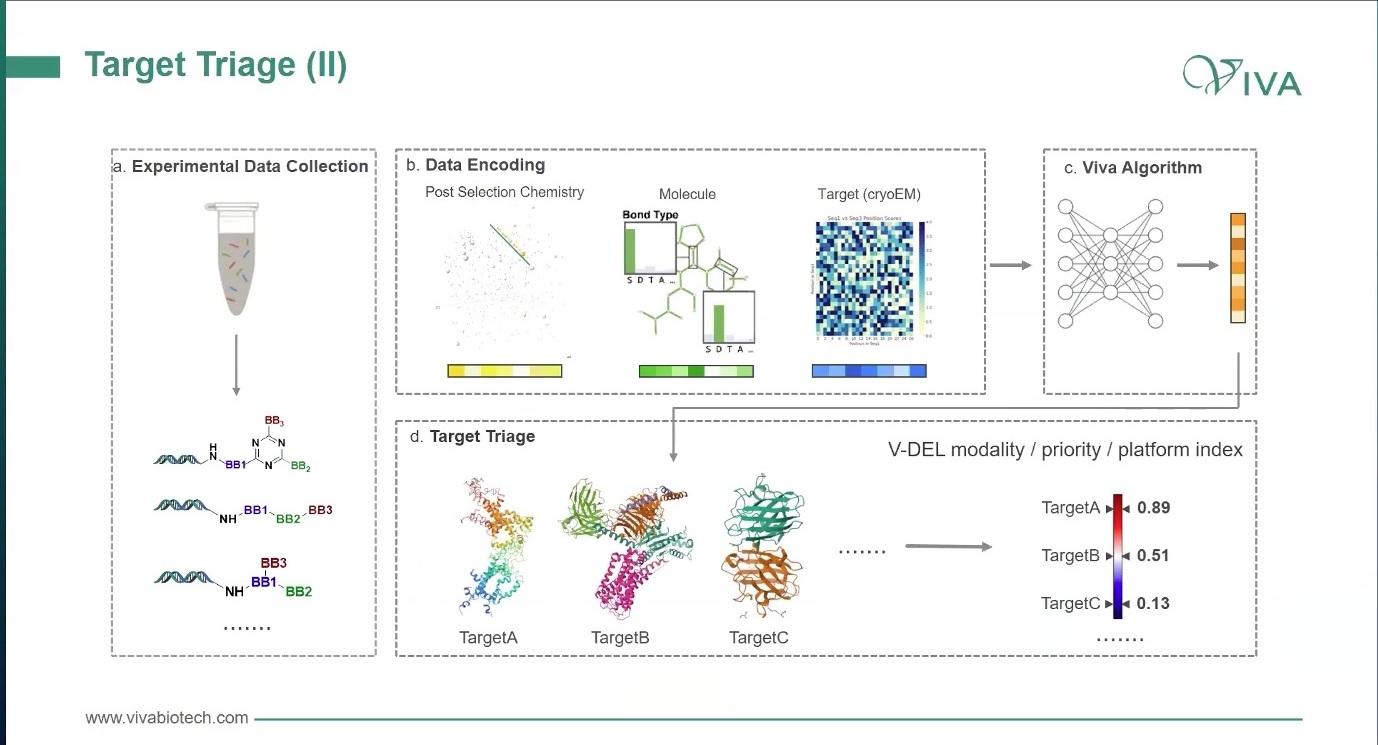
图6. target / modality / platform index 的意义,是把靶点优先级从经验争论推向可比较的数据排序。
对项目来说,这一步越早,代价越低。等到团队已经花了几轮筛选、合成、验证之后,才发现靶点与 modality 不匹配,损失的不只是钱,还有时间、团队注意力和错误叙事的惯性。
Primary hit 不是成果,只是嫌疑人
药物发现中最危险的时刻,未必是没有 hit,而是出现了看起来很漂亮的 primary hit。
一个筛选信号能过 activity threshold,不代表它就是可发展的化学起点。它可能来自化合物不纯,可能来自 aggregate,可能来自 reactive group,可能来自荧光淬灭或光学干扰,也可能只是某类在各种 assay 中反复出现的 frequent hitter。
所以,从 primary hit 到 validated hit,关键不是急着庆祝一个信号,而是把它放进一套验证链里,逐步证明它确实值得继续。

图7. Primary hit validation 的核心是把信号放进一组递进验证:质量确认、干扰排除、正交 assay、选择性、细胞毒性、生物物理结合和竞争实验。
第一步是确认基本质量:LC-MS 纯度是否足够,dose-response 是否合理,IC50 或 EC50 曲线是否有真实浓度依赖。没有这些,后面的讨论都可能建立在噪音上。
第二步是排除明显干扰:PAINS、aggregator、reactive group、fluorescence interference 都不应该被机械地“一票否决”,但它们必须降低团队对这个 hit 的信任度。一个带红旗的分子需要更多证据,而不是更多想象。
第三步是正交验证。换一个 readout,信号是否还在?从 luminescence 换到 TR-FRET,或者从一种检测模式换到另一种检测模式,能不能仍然看到一致趋势?如果换 readout 后信号消失,说明 primary assay 很可能在讲一个假故事。
第四步是反筛和选择性。对 family member 有什么表现?对 unrelated protein 是否也有信号?在细胞体系里是否只是 cytotoxicity 造成的假阳性?这些实验的作用不是锦上添花,而是防止团队把泛活性误读成选择性。
第五步是 biophysical confirmation。功能信号再漂亮,也要回到一个基本问题:分子是否真的与靶点发生 specific binding?SPR、ASMS 或其他 biophysical assay 不能单独证明药效,但可以帮助区分“真实结合”与“体系假象”。
最后,如果有 positive control 或竞争条件,还要进一步看 MOA 是否合理。竞争实验的价值在于把“有信号”推进到“可能以预期方式作用”。
这条验证链背后的原则很简单:validated hit 是被证据逐步放出来的,不是被项目组宣布出来的。
DMTA 的本质,是让错误假设更快暴露
Design-Make-Test-Analyze 看起来像研发流程图里的老词,但它真正的价值不在图,而在节奏。
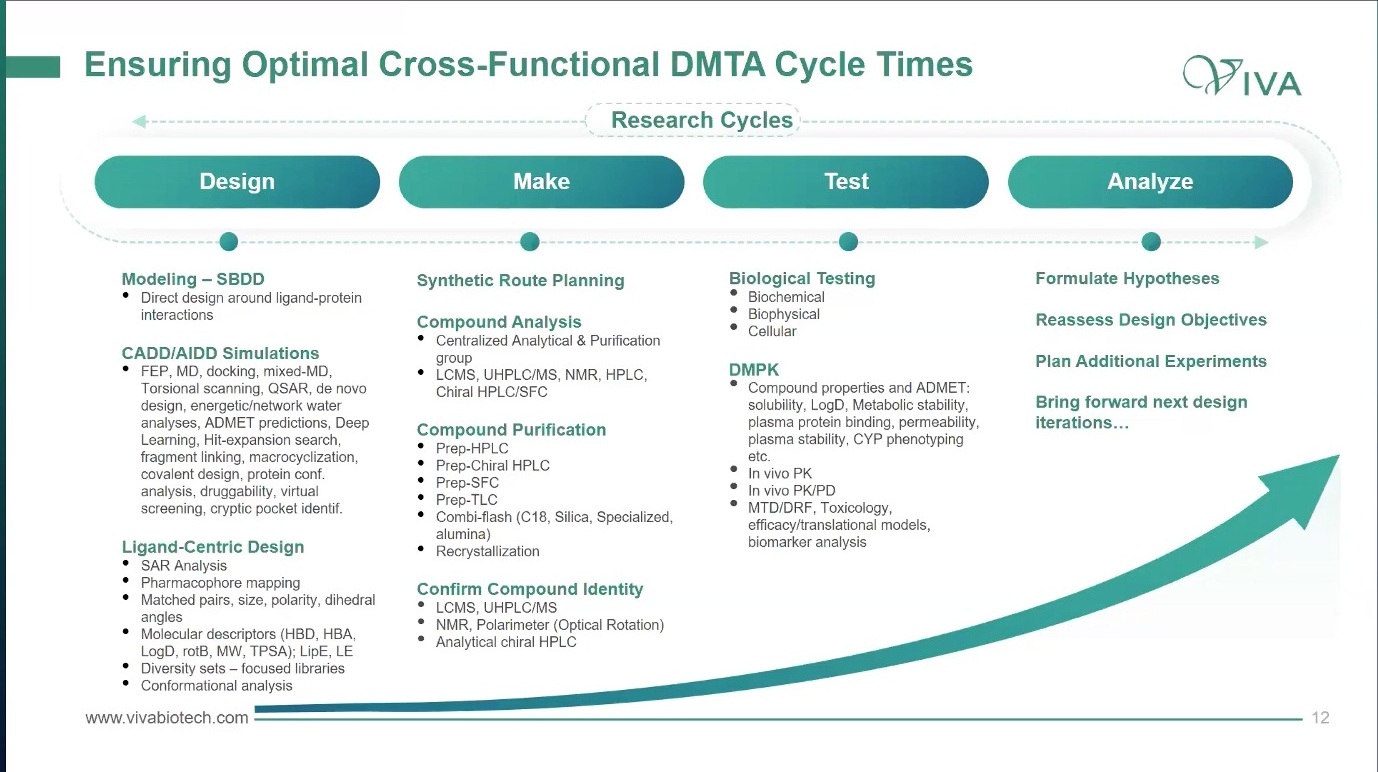
图8. DMTA 的重点不是把 Design、Make、Test、Analyze 写成流程,而是让各职能在同一个 feedback cycle 中更新假设。
一个有效的 DMTA 循环,需要让药化、CADD、合成、生物、DMPK 和项目负责人处在同一个反馈周期里。讨论里提到的理想节奏是一周级,与 primary assay workflow 保持同步。这个细节很关键:如果设计周期、合成周期、测试周期和分析周期不同步,项目会不断堆积过期假设。
Design 阶段不只是画结构。真正有用的 design 要带着 protein model、known interaction、FEP、MD、docking、QSAR、matched pair、descriptor 和 efficiency metric 进入讨论。Make 阶段也不是照单合成,而是根据 synthetic route、ease of synthesis、purification 和 identity confirmation 重新排列优先级。Test 阶段不能只看 potency,还要让 biochemical、biophysical、cellular 和 DMPK 数据一起进入判断。Analyze 阶段的任务则是重新形成假设:下一轮到底该补 SAR、改属性、换 scaffold、补实验,还是停掉整个 design set?
这里有一个很实用的做法:每个 design set 先优先合成 2 到 4 个化合物。如果数据不好,就有机会取消整个 set,而不是让合成团队继续生产一组已经失去意义的分子。合成团队也可以把容易做、且能补足 SAR 的邻近化合物加入队列。
这就是“快”的真正含义。快不是少做实验,也不是压缩判断;快是让错误假设少占用一轮、两轮、三轮资源。
AI 如果要在这里产生真实价值,也必须服务于这个目标。它可以帮助 modeling、generation、多目标优化和优先级排序,但最终仍要回到 DMTA:它是否让下一轮分子更值得合成?是否让下一轮实验更能区分假设?如果不能,AI 只是把猜测包装得更漂亮。
从 hit 到 PCC:活性只是故事的第一章
很多失败不是因为团队找不到活性,而是因为团队太晚才发现活性之外的问题。
从 hit 到 PCC 的 screening cascade,本质上是一种 molecule story telling。一个 compound 不能只讲“我有活性”,还要用一层层数据把自己的故事讲完整:它如何结合靶点,如何进入细胞或体内,如何获得足够暴露,如何避开明显安全性和可开发性风险。第一层是 primary binding / biochemical assay、cellular or biochemical selectivity assay 和 secondary cell-based assay。它们回答的是:有没有活性?这个活性是否和目标相关?
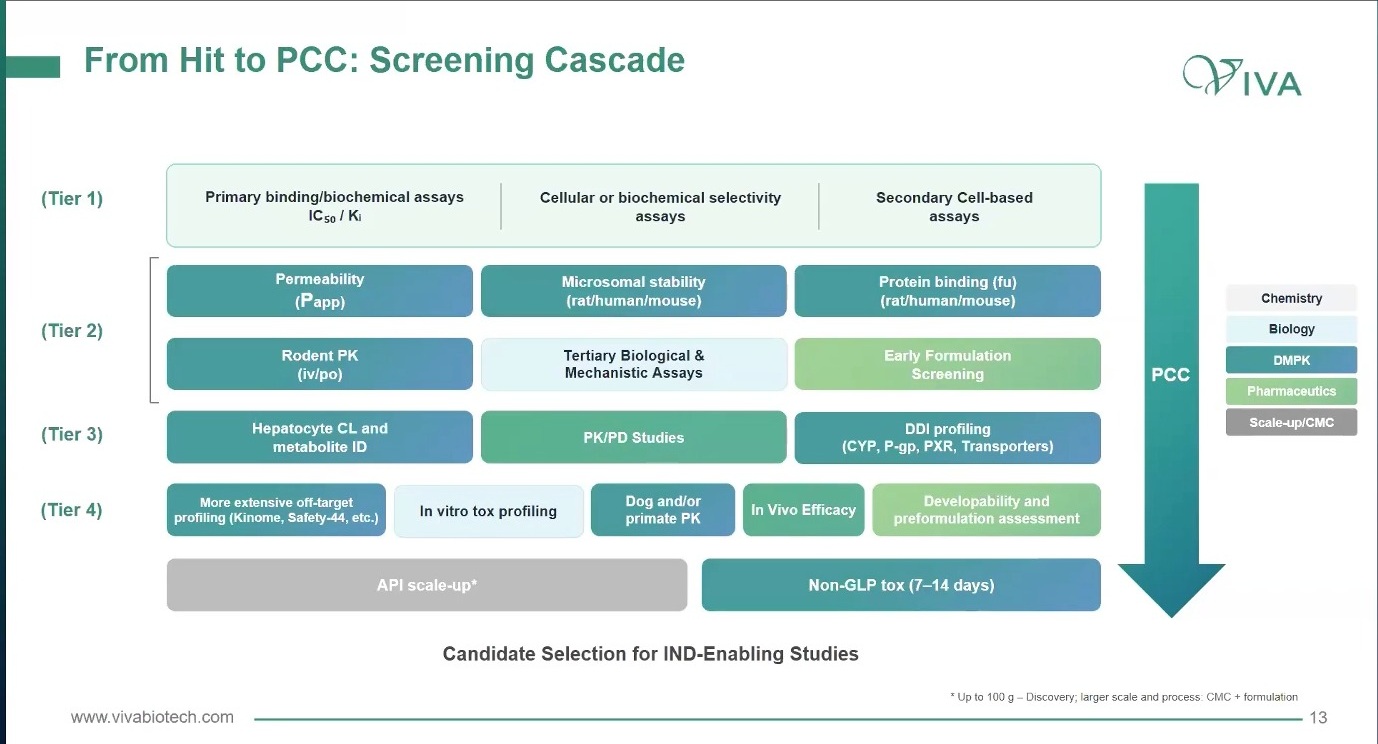
图9. 从 hit 到 PCC 的 cascade 把活性、选择性、DMPK、PK/PD、tox、formulation 和 scale-up 逐层纳入同一个分子故事。
第二层开始进入 compound 在身体里的命运:permeability、microsomal stability、protein binding、rodent PK、mechanistic assay、early formulation。它们回答的是:这个分子能不能到达该到达的地方?是否有足够暴露?是否有基本可开发性?
第三层把药效、暴露、代谢和相互作用连起来:hepatocyte clearance、metabolite ID、PK/PD、DDI profiling,包括 CYP、P-gp、PXR 和 transporter。这个阶段开始逼项目回答更难的问题:体外 potency 能否转化为体内 target modulation?代谢和相互作用会不会提前限制窗口?
第四层靠近候选物选择:off-target profiling、kinome 或 Safety-44、in vitro tox、dog 或 primate PK、in vivo efficacy、developability、preformulation、API scale-up、non-GLP tox。到这里,项目已经不能只讲“这个分子很强”,而要讲“这个分子为什么还能继续”。
PCC 不是最强活性的分子,而是跨过多维风险后仍然成立的分子。
这也解释了为什么早期 tool compound 和 in vivo 信息有时很重要。早期 in vivo 不一定是为了立刻证明临床潜力,而是为了校准后续优先级。如果 target modulation 已经达到预期却没有 efficacy,团队要重新审视 biology、模型、biomarker 或机制,而不是继续盲目追 potency。
Go/no-go 是研发纪律,不是流程装饰
最容易被忽视的复杂性,是组织心理。
一个团队越想要某个 target,越容易让项目无限延长。每一次失败都可以被解释为“实验还不对”“scaffold 还不对”“assay 还不对”“PK 还不够好”。这些解释有时是真的,但如果没有预设 hard stop,它们也会变成项目自我欺骗的语言。
Go/no-go 标准的作用,就是在情绪进入判断之前,先定义什么叫继续、什么叫改变路线、什么叫停止。
常见 hard stop 包括:关键 cellular assay 无法建立;关键 protein construct 无法生产;first-in-class 项目中已经实现最大 target modulation,却没有 efficacy;therapeutic index 过窄且无法通过 selectivity 或机制解释改善;scaffold-specific tox 无法通过 backup series 解决;hit 无法证明 tractability,例如 potency 没有可移动性、binding 非特异、pocket 太浅、selectivity 不足以解释 efficacy。
到了 lead optimization,停损标准会更加现实:DDI、tox panel、cross-species PK、dose linearity、CMC、formulation 和 developability,任何一个都可能改变项目命运。
Go/no-go 不是让团队保守,而是让团队诚实。继续不是因为还有想法,而是因为下一个实验仍然能回答一个值得回答的问题。
组织距离也是研发变量
一体化还有一个容易被低估的维度:组织距离。这里的距离不只是物理距离,而是一个判断从发现问题到影响下一步实验之间,要经过多少交接、等待和翻译。
药物发现里,很多延迟并不是实验本身造成的,而是问题在团队之间流动得太慢。Protein team、screening team、structure team、med chem team、DMPK team 和项目负责人如果处在不同系统、不同会议节奏、不同优先级里,一个简单判断就可能被拖成长链条:蛋白构建是否要改?筛选条件是否要换?某个 readout 是否可信?某个 scaffold 是否值得补 SAR?这些问题本身不一定复杂,但交接会放大复杂性。
一体化真正有价值的地方,是缩短反馈距离。不是因为所有能力放在一起就天然更科学,而是因为它让问题更快回到能解决它的人手里:protein construct 的修改可以直接影响下一轮筛选;结构信息可以直接改变下一组分子设计;DMPK 结果可以及时改变药化优化目标。关键不是“谁拥有更多平台”,而是这些平台之间是否能快速形成共同判断。
这不是说所有项目都必须内部化,也不是说一体化服务商一定优于分散式合作。真正的判断标准应该是:组织结构是否减少了关键问题的等待时间,是否减少了信息在团队之间翻译时的损耗,是否让每一轮实验更快指导下一轮决策。
结语:早期发现的核心能力,是更早知道什么不值得做
如果把这场讨论压缩成一句话,我会说:现代药物发现的效率,不是来自筛到更多 hit,而是来自更早知道哪些 hit、哪些靶点、哪些假设不值得继续。
这听起来不够振奋,但它更接近真实研发。
一个好项目,不是每一轮都有漂亮数据,而是每一轮都能减少不确定性。Hit ID 决策树减少平台选择的不确定性;modality triage 减少路线选择的不确定性;hit validation 减少假阳性的不确定性;DMTA 减少设计假设的不确定性;screening cascade 减少候选物风险的不确定性;go/no-go 减少组织自我欺骗的不确定性。
复杂性不会消失。能做的,是把它前置、拆开、验证,并放进一个有节奏的循环里。
真正的一体化,也不是把所有能力写在一页 slide 上,而是让每个实验都能指导决策。
参考资料
- Viva BioInsights 圆桌讨论:Managing Complexity in Modern Drug Discovery: A Practical Approach to Integrated Screening & Chemistry.
- Viva Biotech 官方资料:About Viva Biotech
- DEL 背景文献:Brenner S, Lerner RA. Encoded combinatorial chemistry. PNAS, 1992.
- Assay interference 背景:Assay Artifacts and Interferences, NCBI Bookshelf.
- Orthogonal/counterscreen 背景:Interferences with Luciferase Reporter Enzymes, NCBI Bookshelf.
- PAINS 背景:Baell JB, Holloway GA. New Substructure Filters for Removal of PAINS. Journal of Medicinal Chemistry, 2010.