一个分子,两套销毁系统:当分子胶开始给抗性留后手
如果把靶向蛋白降解想象成一次“押送”,传统 PROTAC 和分子胶的目标都很直白:把一个本不该见面的靶蛋白和一个 E3 泛素连接酶拉到一起,让细胞自己的泛素-蛋白酶体系统把靶蛋白处理掉。
作者按
如果你当年听到 CRBN/VHL 双 E3 heterotrivalent PROTAC 时,第一反应是“这是不是在玩多价积木”,这个判断并不丢人。把 CRBN、VHL 和靶标配体塞进一个更大的 PROTAC,确实像是在把化学生物学的概念边界往外推;漂亮,但也沉重。真正让这篇 Nature Chemical Biology 变得锋利的是:同一个双 E3 设想,这一次被压缩进了一个 monovalent molecular glue。它不是靠大分子硬接两条 E3 路,而是用一个小分子在 SMARCA2/4、DCAF16 和 FBXO22 之间打开可调界面。于是问题从“能不能玩出双 E3”变成了“能不能把双 E3 做成一种药物设计原则”。
这也是我愿意对这篇文章多给一点情绪的原因。TPD 领域有时太迷恋“把蛋白拖去垃圾桶”这句漂亮话,仿佛只要找到一个 E3,故事就结束了。但癌细胞最擅长的,恰恰是拆掉这种单点依赖:E3 表面、表达量或复合物接口一变,药物就可能从“降解机器”退化成“普通结合物”。这篇论文不是临床“防抗性”的证明;但它确实把 degrader 的问题从“降得够不够低”推进到“失败路径能不能被设计得更难”。
核心要点
- 这篇论文报告了一个单价分子胶 degrader,可并联招募 DCAF16 与 FBXO22 两个不同 E3 ligase substrate receptor 来降解 SMARCA2/4。
- 它和此前 CRBN/VHL heterotrivalent PROTAC 的区别在于:这次不是把两套 E3 recruiter 硬接到更大的 PROTAC 里,而是通过一个 monovalent molecular glue 的诱导界面调动两个 E3。
- 对 compound 1 来说,DCAF16 是更强的主力路径,FBXO22 是功能性备份;单 KO 不能完全阻断降解,双 KO 才能完全救回。
- DMS、完整蛋白质谱和 3.47 A cryo-EM 结构把 DCAF16 侧机制锁定到 C173 共价修饰。
- 极小的化学结构变化可以把 E3 偏好从 DCAF16 推向 FBXO22;DCAF16 的 L59W 突变又能把 DCAF16 依赖重新增强。
- 这不是临床“抗性免疫”的证明,但它提出了一个重要设计原则:degrader 可以被设计成不那么容易被单一 E3 逃逸击穿。

如果把靶向蛋白降解想象成一次“押送”,传统 PROTAC 和分子胶的目标都很直白:把一个本不该见面的靶蛋白和一个 E3 泛素连接酶拉到一起,让细胞自己的泛素-蛋白酶体系统把靶蛋白处理掉。
PROTAC 通常像一根双头绳,一端抓靶蛋白,一端抓 E3。分子胶则更像一枚小楔子:它本身只有一个小分子,却能诱导两种蛋白形成新的接触界面。过去这个故事大多有一个默认前提:一个 degrader 对应一个主要 E3。这个前提让机制清楚,也让药化优化有抓手,但它也留下一个显眼风险:如果肿瘤细胞把这条 E3 路径改坏了,药物的降解能力就可能被拆掉。
Valentina A. Spiteri、Dmitri Segal、Alejandro Correa-Sáez 等人在 Nature Chemical Biology 发表的新论文,把矛头对准的正是这种单一路径依赖。作者研究的是 SMARCA2/4,一组 BAF/SWI/SNF 染色质重塑相关蛋白,也是肿瘤药物发现中长期受关注的染色质调控脆弱性。论文中的关键分子 compound 1 不是常规双功能 PROTAC,而是一个单价分子胶:它带有 SMARCA2/4 bromodomain 结合部分,以及一个 propargyl-azepane degradation tail。
异常出现在找 E3 的时候
一开始,数据看起来像是熟悉故事。compound 1 能在 HEK293T 和 HCT-116 细胞中降解 SMARCA2/4;定量蛋白质组学显示主要下降的是 SMARCA2/4 和 PBRM1;去掉关键 tail 的近似物 1* 不再降解;抑制 UBA1、neddylation 或蛋白酶体都能阻止 SMARCA2 降解。这说明它确实在利用 CRL 依赖的泛素-蛋白酶体系统。
真正的异常出现在找 E3 的时候。
CRISPR 筛选把 DCAF16、DDB1、RBX1 推到前面,看上去 DCAF16 就是答案。但 DCAF16 敲除并不能完全救回 SMARCA2 降解。作者继续做近邻标记蛋白质组学,发现 FBXO22 也被 compound 1 诱导靠近 SMARCA2。随后单 KO 和双 KO 的实验把机制钉住:单独敲掉 DCAF16 或 FBXO22,只会减慢或削弱降解;同时敲掉两者,才会完全阻断 SMARCA2/4 的泛素化和降解。
这就是这篇论文最关键的一步:compound 1 不是换了一个 E3,而是并联了两个 E3。更准确地说,它同时利用 CRL4-DCAF16 和 CRL1-FBXO22。TR-FRET 进一步证明 compound 1 能在体外诱导 SMARCA2 bromodomain 与两套 CRL 复合物形成三元复合体,只是 DCAF16 更强,EC50 为 110 nM,而 FBXO22 的 EC50 大于 1 uM。在 SMARCA4-HiBiT 细胞实验里,compound 1 的 WT DC50 达到 2.2 nM。
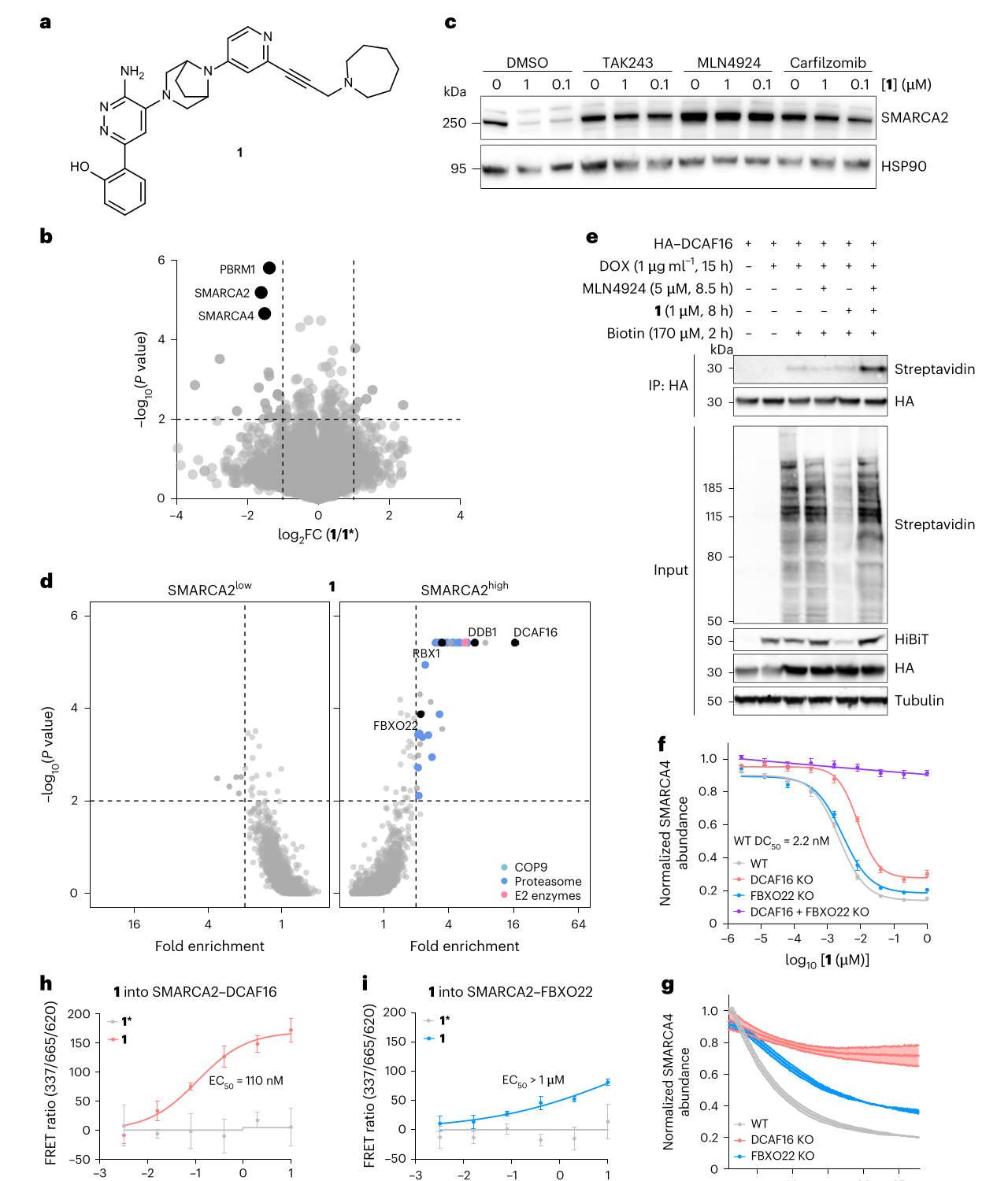
图源:Spiteri et al., Nature Chemical Biology, 2026;裁自论文 PDF,CC BY 4.0。图 1 展示 compound 1 的结构、蛋白质组学选择性、CRISPR 筛选、DCAF16 近邻标记、KO 降解曲线和 DCAF16/FBXO22 体外三元复合体形成。
| 问题 | 论文给出的答案 |
|---|---|
| compound 1 是否真在走泛素-蛋白酶体系统? | 是。UBA1、neddylation、蛋白酶体抑制都能阻止降解。 |
| 只依赖 DCAF16 吗? | 否。DCAF16 KO 不能完全救回。 |
| FBXO22 是旁观者吗? | 否。近邻标记、CRISPR 弱命中、KO 和 TR-FRET 都支持其参与。 |
| 哪个 E3 更强? | compound 1 下 DCAF16 更强,是 primary driver;FBXO22 是 secondary contributor。 |
| 双 E3 是否有功能冗余? | 是。单 KO 不足以完全阻断,DCAF16/FBXO22 双 KO 才能完全 ablate 降解。 |
作者把 DCAF16 这条路径的分子动作看清楚了
“两个 E3”如果只停在细胞遗传学层面,仍然像一个黑箱。论文最漂亮的部分,是作者把 DCAF16 这条路径的分子动作看清楚了。
他们对全长 DCAF16 做深度突变扫描,一共覆盖 4,300 个变体,找到了关键位点 C173。C173S 会破坏 compound 1 介导的 SMARCA2 降解;完整蛋白质谱显示 compound 1 会共价标记野生型 DCAF16,却不能标记 C173S 突变体。换句话说,这个分子不是只靠“贴近”,而是在 DCAF16 的 C173 上形成了共价接触。
随后 cryo-EM 给出了 3.47 A 的三元复合体结构:SMARCA2 bromodomain、compound 1、DCAF16:DDB1:DDA1 被固定在一起。结构中,DCAF16 上原本未解析的 E164-C177 back loop 被看见了,C173 就藏在这段 loop 里,并与 compound 1 形成连续电子密度。compound 1 的 pyridine 与 DCAF16 W181 形成 pi-stacking,azepane 环则嵌入由 SMARCA2 Q1411-L1412 与 DCAF16 P170-S172/Y62 共同构成的口袋。这个新界面埋藏了约 1,057 A2 表面积。
这不是一个“胶水随机糊住蛋白”的故事,而是一套精细到残基级别的诱导接触。
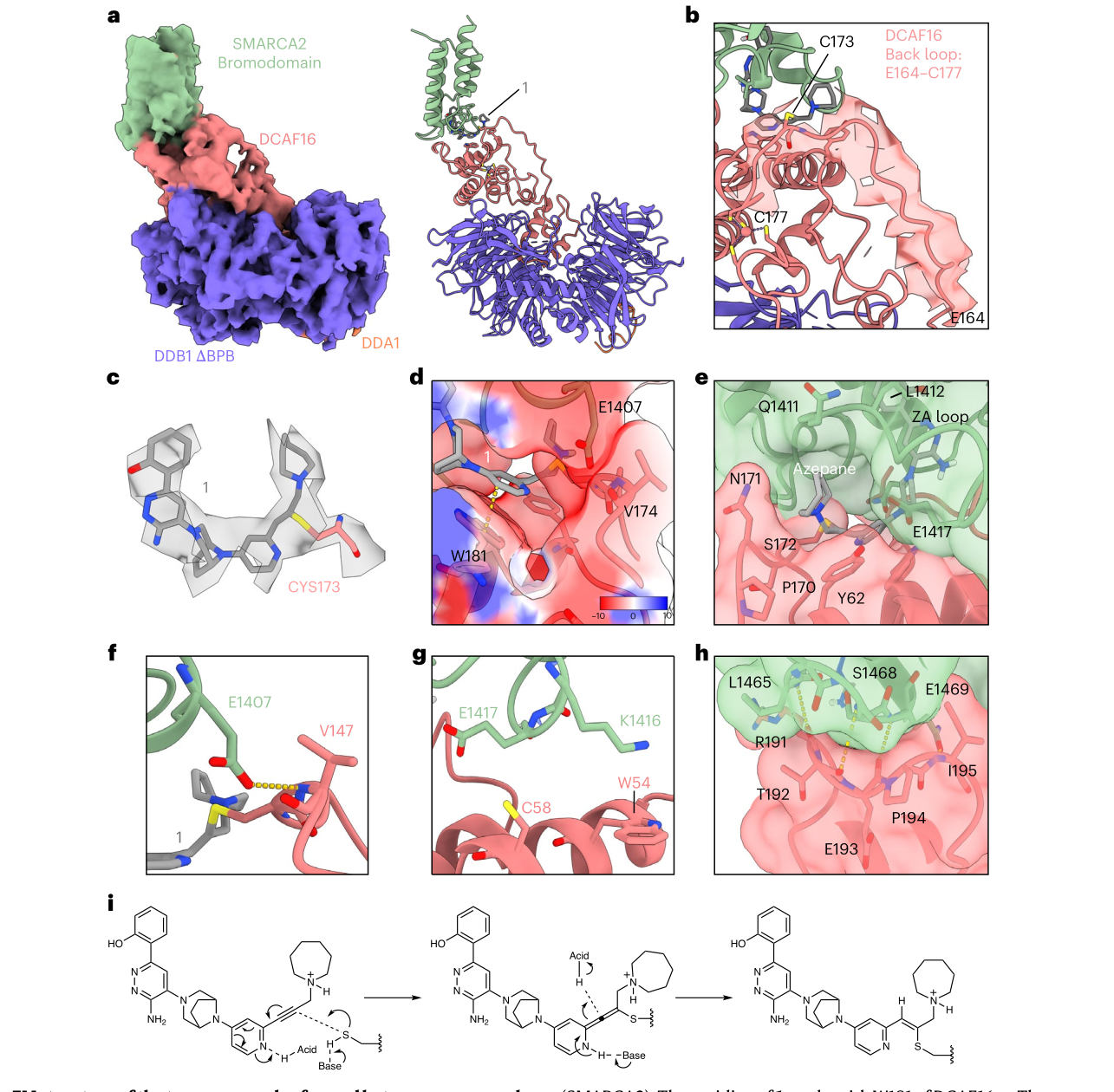
图源:Spiteri et al., Nature Chemical Biology, 2026;裁自论文 PDF,CC BY 4.0。图 3 把 C173 所在的 DCAF16 back loop、compound 1 的共价连接和 SMARCA2-DCAF16 接触界面放到同一个结构框架里。
可调,而不是偶然
更有意思的是,这套机制不是固定死的。compound 2 只是在 tail 上做了小改动,却把主力 E3 从 DCAF16 推向 FBXO22。对 compound 2 来说,FBXO22 KO 足以阻断 SMARCA4 泛素化和 SMARCA2 降解;SMARCA4-HiBiT 里 WT DC50 为 73 nM。
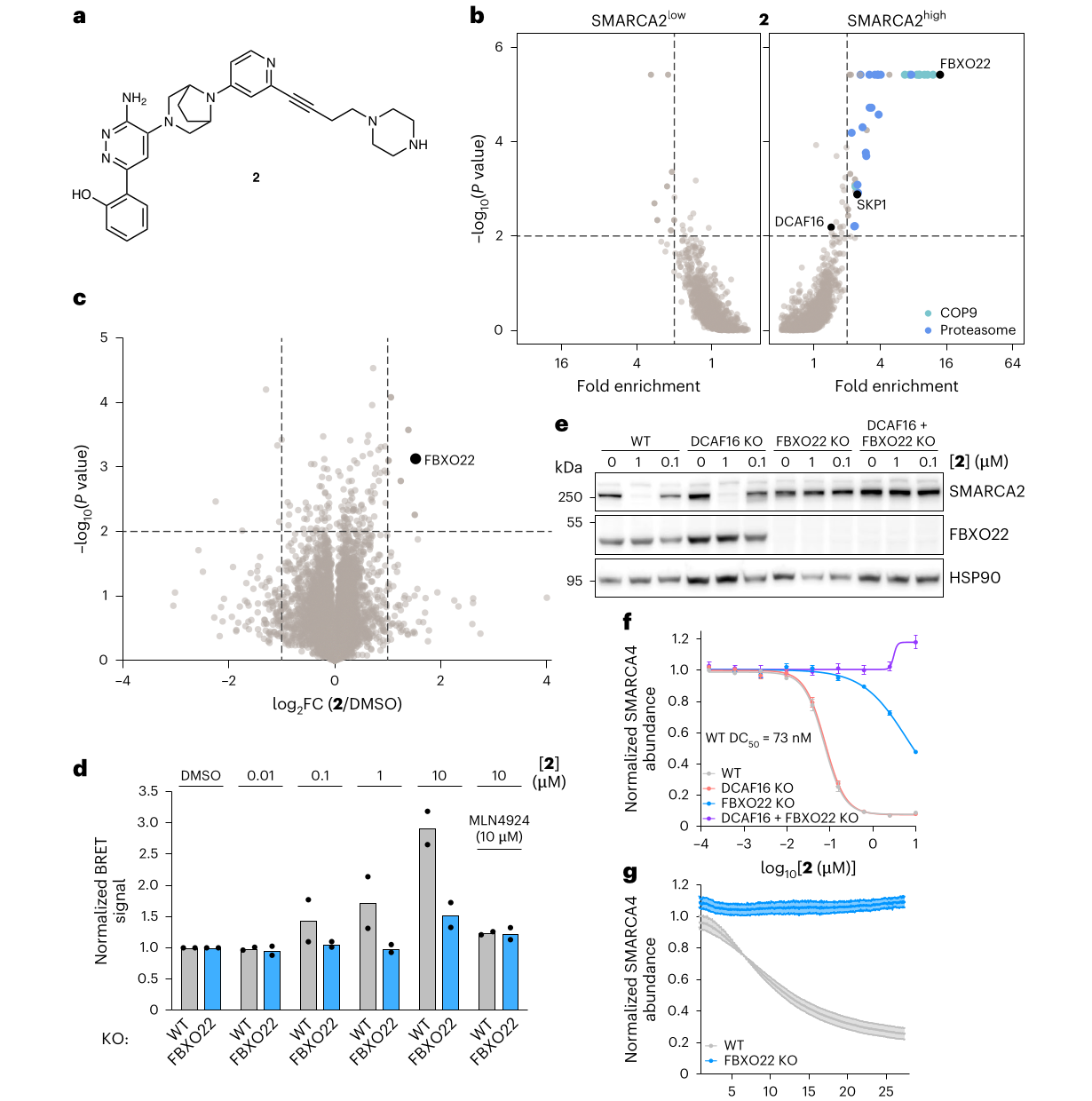
图源:Spiteri et al., Nature Chemical Biology, 2026;裁自论文 PDF,CC BY 4.0。图 4 是“偏好可调”的关键证据之一:compound 2 的 CRISPR/BioID/KO/HiBiT 结果都指向 FBXO22 成为主导路径。
compound 3,也就是相关文献中的 G-6599,与 compound 1 几乎只差一个 methylene,也表现出更偏向 FBXO22 的 driver ligase switch,同时仍保留双 E3 依赖的影子。反过来,compound 4 和 5 的小改动会直接丢失降解活性,说明这里不是“随便修饰都能切换”,而是一个狭窄但真实的可调窗口。
作者还从遗传方向证明了同一件事。DCAF16 的 L59W 突变可以增强 compound 2 和 3 对 DCAF16 的依赖。TR-FRET 中,在 compound 2 存在时,DCAF16-L59W 与 SMARCA2 形成复合体的 EC50 为 0.31 uM,而野生型 DCAF16 为 1.98 uM;完整蛋白质谱中,compound 2 对 DCAF16 的标记比例也从 WT 的 6.3% 提高到 L59W 的 33.7%。作者提出的结构解释很直观:W59 可能帮助稳定 W181 的构象,形成一个更有利于 pyridine 堆叠的 “WW-pyridine shelf”。
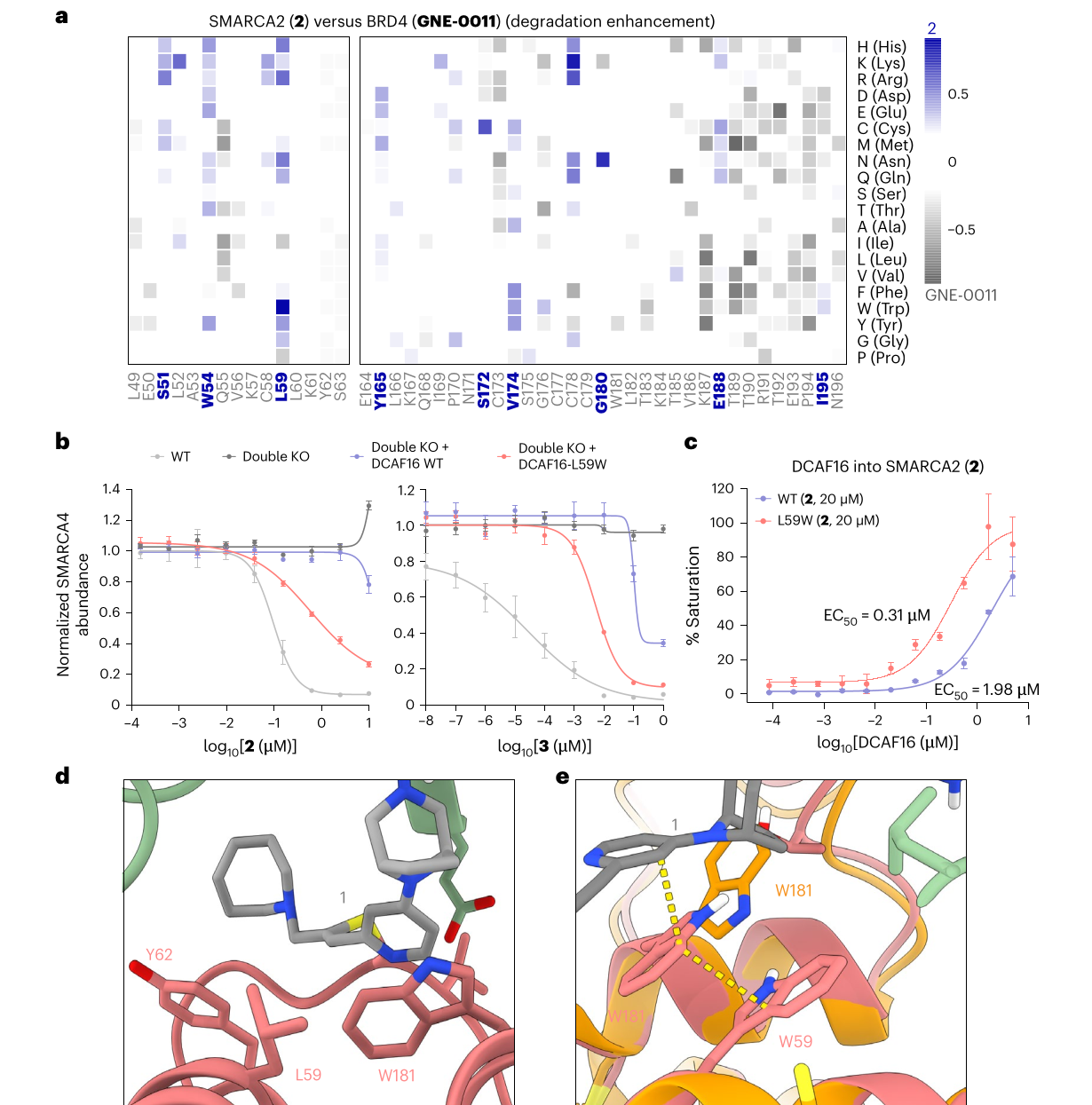
图源:Spiteri et al., Nature Chemical Biology, 2026;裁自论文 PDF,CC BY 4.0。图 5 展示同一个结论的遗传侧证据:改变 E3 表面,也能改变 degrader 的 E3 偏好。
| 调节方式 | 结果 | 设计含义 |
|---|---|---|
| compound 1 | DCAF16 为主,FBXO22 参与 | 同一分子可并联两条 E3 路径 |
| compound 2 | driver 偏向 FBXO22 | tail 的小改动可重排 E3 依赖 |
| compound 3 / G-6599 | 更偏 FBXO22,但仍有双 E3 逻辑 | 一个 methylene 也可能改变 ligase preference |
| compound 4/5 | 失去降解 | 可调窗口狭窄,不能把 SAR 简化成“随便微调” |
| DCAF16 L59W | 增强 DCAF16 对 2/3 的参与 | E3 表面本身也可以改变 degrader 偏好 |
这就是“tunable”的真正含义:E3 选择性不是实验偶然,而可以被小分子结构和 E3 表面共同调节。
为什么药化设计人员会兴奋
这对药化设计人员的诱惑很大。过去我们常问:“这个 degrader 能不能把靶蛋白降下去?”这篇论文逼着我们问另一个问题:“当细胞试图逃逸时,它需要破坏几条路?”
如果一个分子只依赖一个 E3,抗性可以集中打击一个接口;如果一个分子能并联两个 E3,那么单一路径的损失就不一定足以完全取消降解。通讯作者在社媒中把它称作 built-in molecular backup,这个说法有传播上的锐度,也基本抓住了机制重点。
更深一层的意义是:degrader 的优化目标不必只剩 potency、selectivity 和 PK。E3 dependency profile 本身也可以成为设计参数。你可以问:这个分子主要靠哪条 E3 路?备用路有多强?如果主力 E3 降低表达,备用路能否补上?如果某个肿瘤背景中 FBXO22 更可靠,能否把分子推向 FBXO22?如果 DCAF16 路径更安全,能否把结构拉回来?
这比“又一个降解剂”更重要。它把 degrader 的失败模式纳入设计空间。
必须保持冷静的地方
冷水也必须泼在这里。
第一,这篇论文没有证明临床层面的 resistance-proof。它证明的是一个机制框架:双 E3 并联和偏好调谐有可能提高抗性韧性。
第二,这套分子带有共价反应逻辑,反应性、选择性、毒性和组织背景都需要继续被拆开。
第三,SMARCA2/4 的肿瘤治疗价值依赖具体遗传背景,不能简单写成“所有癌症靶点”。
第四,蛋白质组学中 PBRM1 也被降解。这可能是合理的同源 bromodomain 结果,也可能在不同应用场景中成为需要管理的选择性问题。
但这些限制并不削弱论文的概念价值。相反,它们让这项工作更像一个真正有用的设计原则,而不是一次社媒式胜利宣言。
最后真正该记住的事
最值得记住的一句话也许不是“一个分子降解了 SMARCA2/4”,而是:一个分子可以把同一个靶蛋白交给两套不同的 E3 处理,而且这两套系统的权重可以被调。
对 TPD 来说,这意味着药物设计开始触碰一个更高级的问题:不是怎样把蛋白降得更低,而是怎样让降解这件事更不容易被细胞拆掉。
主要来源
- Spiteri, V. A., Segal, D., Correa-Sáez, A. et al. “Dual E3 ligase recruitment by monovalent degraders for tunable SMARCA 2/4 degradation.” Nature Chemical Biology, published 12 May 2026. https://www.nature.com/articles/s41589-026-02224-y
- Local PDF used for close reading:
/Users/fanyi/Downloads/s41589-026-02224-y.pdf - User-provided corresponding-author social media text and lecture-context note in the prompt.
- Bond, A. G. et al. “Leveraging Dual-Ligase Recruitment to Enhance Protein Degradation via a Heterotrivalent Proteolysis Targeting Chimera.” Journal of the American Chemical Society, 2024. https://pubmed.ncbi.nlm.nih.gov/39606859/
- Farnaby, W. et al. “BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design.” Nature Chemical Biology, 2019. https://www.nature.com/articles/s41589-019-0294-6
- Villemure, E. et al. “Rational design of potent small-molecule SMARCA2/A4 degraders acting via the recruitment of FBXO22.” Nature Communications, 2025. https://www.nature.com/articles/s41467-025-64669-4